0512-6856 7071
info@ecellbio.com
过去,中枢神经系统(CNS)药物研究主要依赖于啮齿动物模型或细胞体外模型等传统方法。由于人类和啮齿类动物间的物种差异,所获得的数据难以真实地模拟神经发育和疾病机制等。随着干细胞技术的发展,培养人大脑类器官成为目前神经科学研究领域炙手可热的研究项目。大脑类器官是模拟人脑的生理特性的独特而绝佳的工具,可用于研究正常脑与疾病脑的建模,用于阐明中枢神经系统疾病的发病机制,亦可用于神经发育疾病的探索,或用作中枢神经系统药物筛选的工具。
脑类器官通常从人多能性干细胞(ESCs/iPSCs)开始培养,自发形成脑发育早期所具备的结构和层次。但脑细胞团簇达到一定尺寸后,可发育的阶段会受到营养缺乏和氧供应的限制,继而神经元开始死亡,结构停止发育。目前,大脑类器官的培养主要分为两种方法,引导分化法 (Guided)和非引导 (Un-guided)分化法。
①非引导分化法依赖于细胞自身的形态发生和内在的分化能力,将外在干扰最小化,得到的类器官有前脑、中脑、后脑、视网膜和脉络丛等多种细胞谱系,该种方法的缺陷在于高可变性和异质性。
②而引导分化法是加入一些外源性的模式因子,诱导hPSCs分化为想要的细胞谱系。
在类器官结构中,多能性干细胞(ESCs/iPSCs)诱导为拟胚体 (Embryoid Body, EB),在外胚层形成后,引导分化为神经或非神经方向。引导分化法获得的类器官依赖分化过程中添加的生长因子,大多数是脑区特异性的,如皮层、海马、中脑、大脑等。引导分化法得到的类器官含有神经祖细胞、神经元、星形胶质细胞及其他的大脑细胞。由于是引导性分化,批次间的可变因素少,可以产生对应比例的特异性细胞类型。很多团队都在尝试用不同的方法来培养类器官,分别培养脑区特异的类器官后再将其共培养,类器官会自我融合形成含有不同脑区的类器官,该模型可以用于研究脑区间的相互作用。

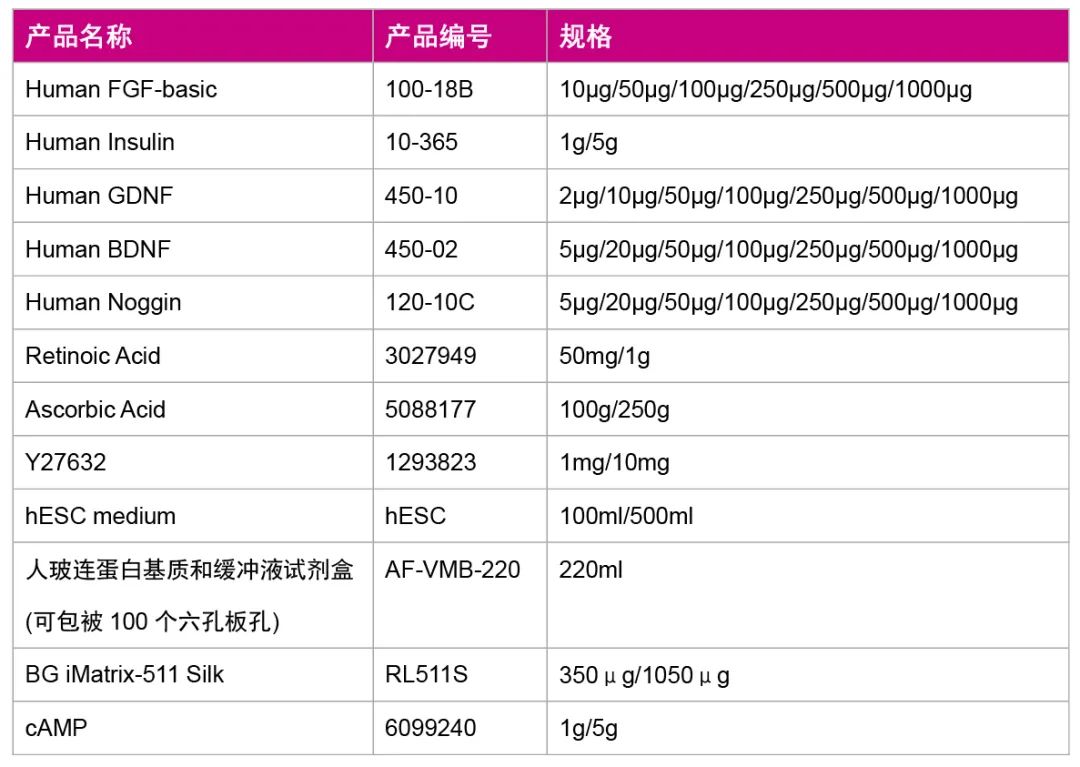
2013年,Lacaster和Knoblich等发表在Nature及Nature protocol上的3D大脑类器官的制作方法使得体外用人多能干细胞 (包括胚胎干细胞及诱导多能干细胞)诱导3D大脑类器官的技术前进了一大步,他们通过使用一种允许类器官获取能量和氧气的新方法克服了这一局限,使用一个微小的振动刀片将类器官切成半毫米切片,然后将它们放在覆盖富含营养的液体的多孔膜上。然后,类器官可同时从上方吸收氧气,从下方吸收营养物质,使其在培养物中继续发育一年。图1是该方法的概括。
|
诱导EB形成1-2h |
1. 当ESCs/iPSCs在六孔板中长到融合度为70-80%时用于诱导EB,通常每个六孔板孔的细胞可用于诱导一整个96孔板。 注:干细胞克隆的形态对于大脑组织形成的成功与否非常关键。克隆需呈现多能性的特征 (如边缘清晰,克隆紧密等),且无分化倾向;
① 用1ml的无钙镁的D-PBS洗涤细胞后,向每孔中加入600μl的含0.5mM的EDTA的无钙镁的D-PBS溶液。培养箱孵育4min; ② 轻柔吸出EDTA溶液,注意不要破坏克隆,加入1ml的Accutase。放回培养箱孵育4min; ③ 用1ml的mTeSR1培养基吹散克隆,使其从培养皿底部脱落。转移2ml至15ml离心管中,用1ml移液器将克隆吹散为浑浊的单细胞悬液; ④ 270g 室温离心细胞5min,同时用台盼蓝检测死细胞,用血细胞仪或自动细胞计数仪细胞计数;检测两次取平均值; ⑤ 用1ml的含50μM的ROCK抑制剂Y-27632的低bFGF的ESCs培养基重悬细胞,吹打,保证细胞呈单细胞重悬的状态。然后,用含Y-7632的低FGF-2的培养基重悬细胞,使细胞浓度为每150μl含9000个活细胞; ⑥ 每个96孔板孔中放入150μl的细胞悬液,置于培养箱继续培养; |
|
饲养EB,早期胚层分化5-7d |
2. 24h后,显微镜下观察,可见有清晰边缘的小的EB形成,边缘有许多死细胞围绕在EB周围,为正常现象,不会干扰EB在中间形成,继续置于37℃的5%二氧化碳的培养箱中培养; 3. 隔天轻柔地吸去半量培养基,避免干扰到EB和底部的细胞,补充150μl的新鲜培养基(总体积会大于150μl,量的准确与否不重),培养基中添加Y-27632(1:100),FGF-2浓度为4ng/ml,直到EB直径大于350-450μm为止,通常,仅前4天需要添加二者; 4. EB直径达到350-600μm时,用3中的培养基隔天饲养EB,培养基中不含Y-27632和FGF-2; |
|
原始神经上皮的诱导4-5d |
5. EB直径达到500-600μm时,边缘开始变得明亮,并且呈现光滑的边缘 (通常为第6天),将每个EB用剪掉枪头的200μl移液器转移至含有500μl的神经诱导培养基的低吸附24孔板中,轻柔且不要破坏EB,继续培养; 注:将200μl移液器枪头剪掉,使开口直径约为1-1.5mm。确保开口不要太小,避免破坏EB,但也不能太大,太大会难以吸去EB。不要试图用刮刀将EB铲出,会破坏EB的结构;
7. 2d后,组织培养显微镜观察EBs,形态边缘变得更加明亮,提示神经外胚层的分化;一旦这些区域开始显示与神经上皮形成一致的假复层上皮的放射状组织,这一般发生在神经诱导培养基中的4-5d后,继续进行第8步,将组织聚集转移到Matrigel液滴中; 注:健康的细胞聚集有光滑的边缘,神经上皮边缘在光学上呈半透明状。有时组织可能在光学上表现为非放射状组织的半透明组织,虽然这不是理想状态,但没有观察到这对类器官形成有长期的负面影响。重要的是,如果在神经诱导中再多放置1-2天,这些非放射区域就会变成放射状,但是如果不及时转移到基质凝胶(步骤8),其他已经放射状的区域可能开始收缩并失去形成神经上皮芽的能力。当有神经外胚层出现时,需尽快将组织转移到Matrigel中,如果太晚,会影响脑组织的晚期形态; |
|
将神经外胚层组织转移至Matrigel液滴中1-2h |
8. 4℃下冰上解冻Matrigel。观察发现,500μl的Matrigel在冰水混合物浴时,1-2h后会恢复液体状态; 9. 将Parafilm膜置于一个空的带有凹坑的中号枪头盒上,将手指按向Parafilm膜形成凹坑,每个坑的体积约为200μl; 注:Parafilm不能高压灭菌,所以不能彻底灭菌,因此需保证Parafilm膜保存在干净的环境中,准备凹坑前,向手套和Parafilm膜喷洒70%乙醇消毒;在这一步也可以在培养基中添加抗生素以避免污染;
11. 用200μl的移液器转移神经外胚层组织,每个凹坑中放置1个; 注:将200μl移液器枪头剪掉,使开口直径约为1.5-2mm。确保开口不要太小,避免破坏EB,但也不能太大,太大会难以吸去EB。不要试图用刮刀将EB铲出,会破坏EB的结构;
13. 每个组织快速滴入Matrigel,每孔约30μl,使Matrigel滴入Parafilm凹坑中; 注:快速滴入Matrigel,避免组织干掉。尽量一次性滴入Matrigel,一次性包埋好16个组织;
15. 将此60mm培养皿置入37℃培养箱中,孵育20-30min以使Matrigel聚合; 16. 取出60mm培养皿,加入5ml不含Vitamin A的脑类器官分化培养基; 17.将Parafilm膜反过来并摇动培养皿,直至Matrigel液滴从凹坑中滴入培养基中;不容易脱落的液滴可以用镊子用力晃动Parafilm膜使其脱落,将培养皿置于培养箱中继续培养组织。 |
|
神经上皮芽的固定培养 4d |
18. 24h后,观察包埋的组织,1-3d内,组织会呈现包含液体空腔的进一步扩张的神经上皮样; 注:从Matrigel主体中会迁移出一些细胞类型,通常呈成纤维细胞样,这些细胞代表了非神经特性的群体,从神经诱导时逃脱出来;然而这些细胞通常不能生存,且迁移出来后,对神经上皮芽的形成呈现促进作用;
注:换液时,倾斜培养皿使液滴下沉,轻柔吸出培养液。尽量在不破坏类器官的情况下,吸出更多的培养液。更换为5ml的新鲜培养液; |
|
脑组织的生长 ~1年 |
20. 4h的静止培养后,用开口约为3mm的剪去头部的1ml移液枪头将包埋好的类器官转移至125ml的震动反应器中,加入75~100ml的含Vitamin A的脑类器官分化培养液,将此生物反应器放置在培养箱中安装的磁搅拌板或轨道振动筛上用85rpm的速度震动。也可以将60mm的培养皿中培养液更换为含Vitamin A的脑类器官培养液,将其置于轨道振动筛上培养; 注:每个生物反应器中不要转移超过2个60mm培养皿的类器官(32个),太多类器官会引起类器官的融合;
轨道振动筛可用于分析多种培养条件,同时使用多种基因突变剂处理,而传统的搅拌板只能同时防止4-6个单独的瓶;用生物反应器和轨道振动筛培养的脑类器官未观测到有区别; 21. 振动筛上的类器官每3-4天全量换液,摇瓶上的类器官每周换液,注意观察形态;在合适的时间点取样本用于实验研究; |
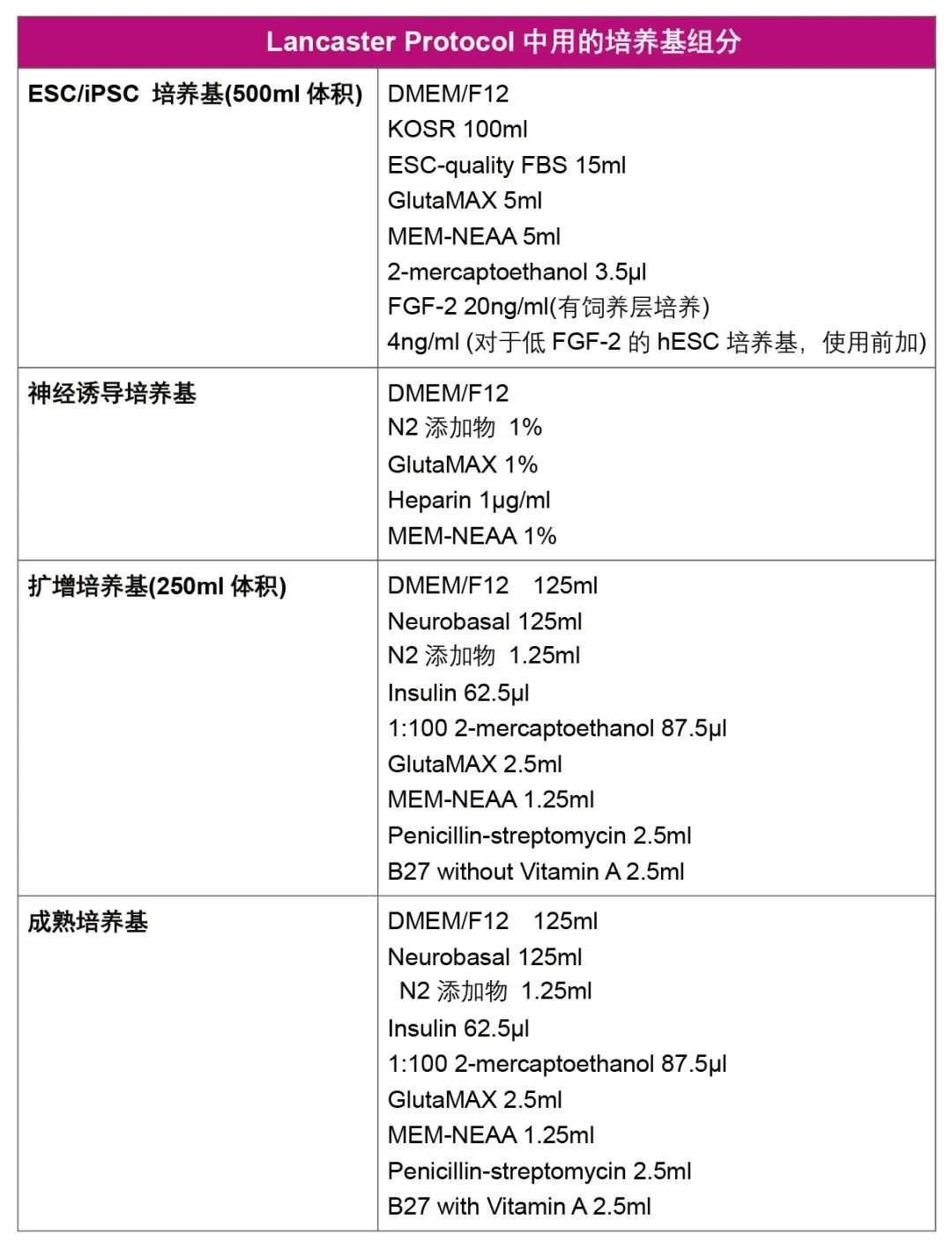
我们提供人脑类器官培养所需的各种细胞因子及小分子,并提供人ESC/iPSCs培养所需的培养基及培养器皿包被基质,产品详情如下表: